SATISFY: A EuroBloodNet Multicenter, Single-Arm Phase 2 Trial of Mitapivat in Adult Patients with Erythrocyte Membranopathies and Congenital Dyserythropoietic Anemia Type II – Results from the 8-Week Dose-Escalation Period
Erythrocyte membranopathies represent a heterogeneous group of diseases characterized by hemolysis resulting from structural and functional defects in cytoskeletal, (trans)membrane and ion-channel proteins. Congenital dyserythropoietic anemia type II (CDA II) is another form of rare hereditary anemia characterized by ineffective erythropoiesis and hemolysis. Current treatment options are mainly supportive. Mitapivat is a first-in-class oral allosteric activator of the pyruvate kinase (PK), a key glycolytic enzyme. Mitapivat enhances glycolysis, subsequently increasing ATP and decreasing 2,3-diphosphoglycerate within the red blood cell. Recent evidence suggests a relatively decreased PK activity in hereditary spherocytosis (HS), attributed to loss of membrane-bound PK due to impaired structural integrity.
This study (NCT05935202) aims to evaluate the safety and efficacy of mitapivat in patients with erythrocyte membranopathies and CDA II. This study is being conducted in the Netherlands and Denmark. Eligible subjects were adults with a genetically supported (ACMG class 3-5), non-transfusion dependent erythrocyte membranopathy or CDA II, with an average hemoglobin (Hb) concentration of <11.0 g/dL for females and <13.0 g/dL for males. Key exclusion criteria were known history of PK deficiency, regularly scheduled blood transfusion episodes, and/or any significant medical condition.
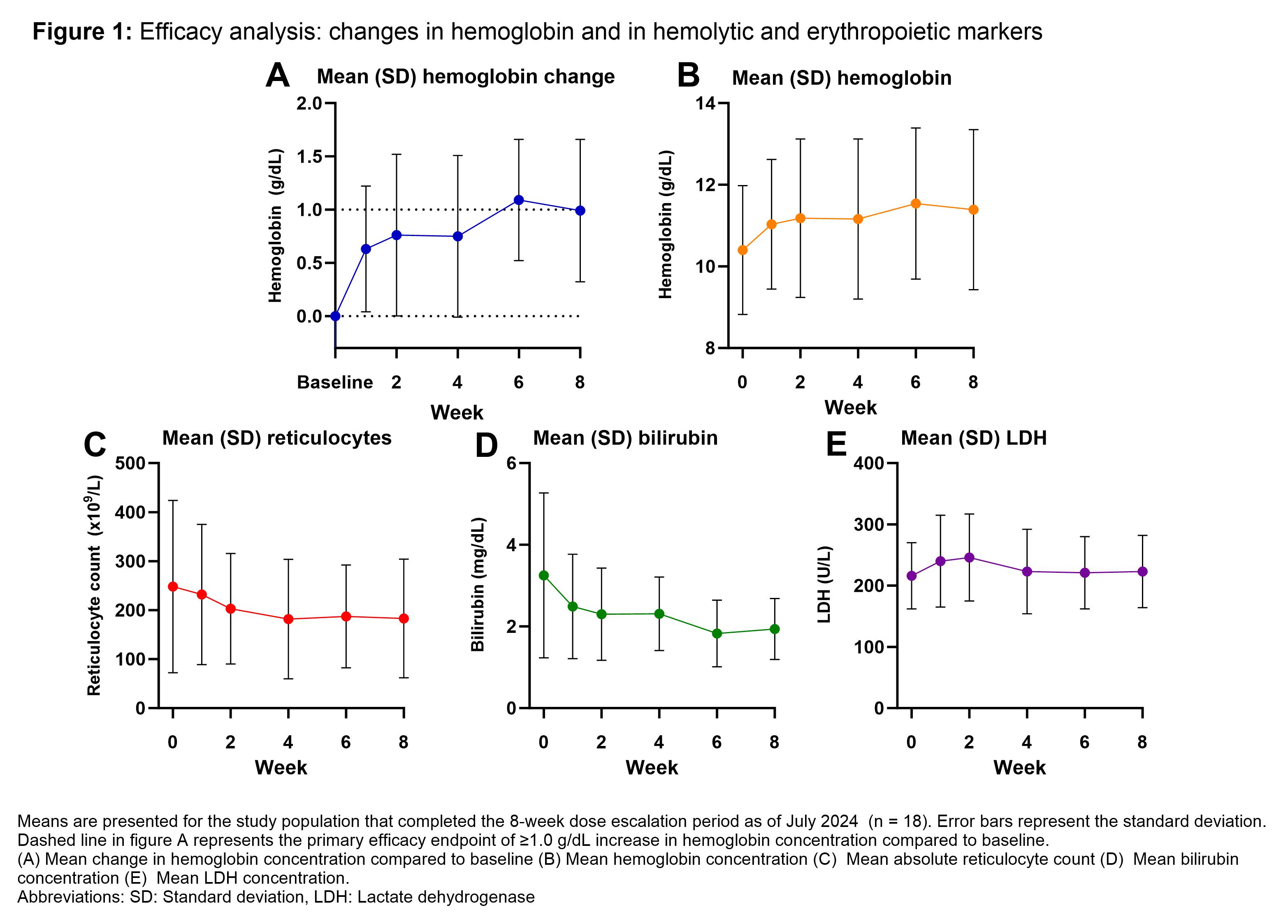
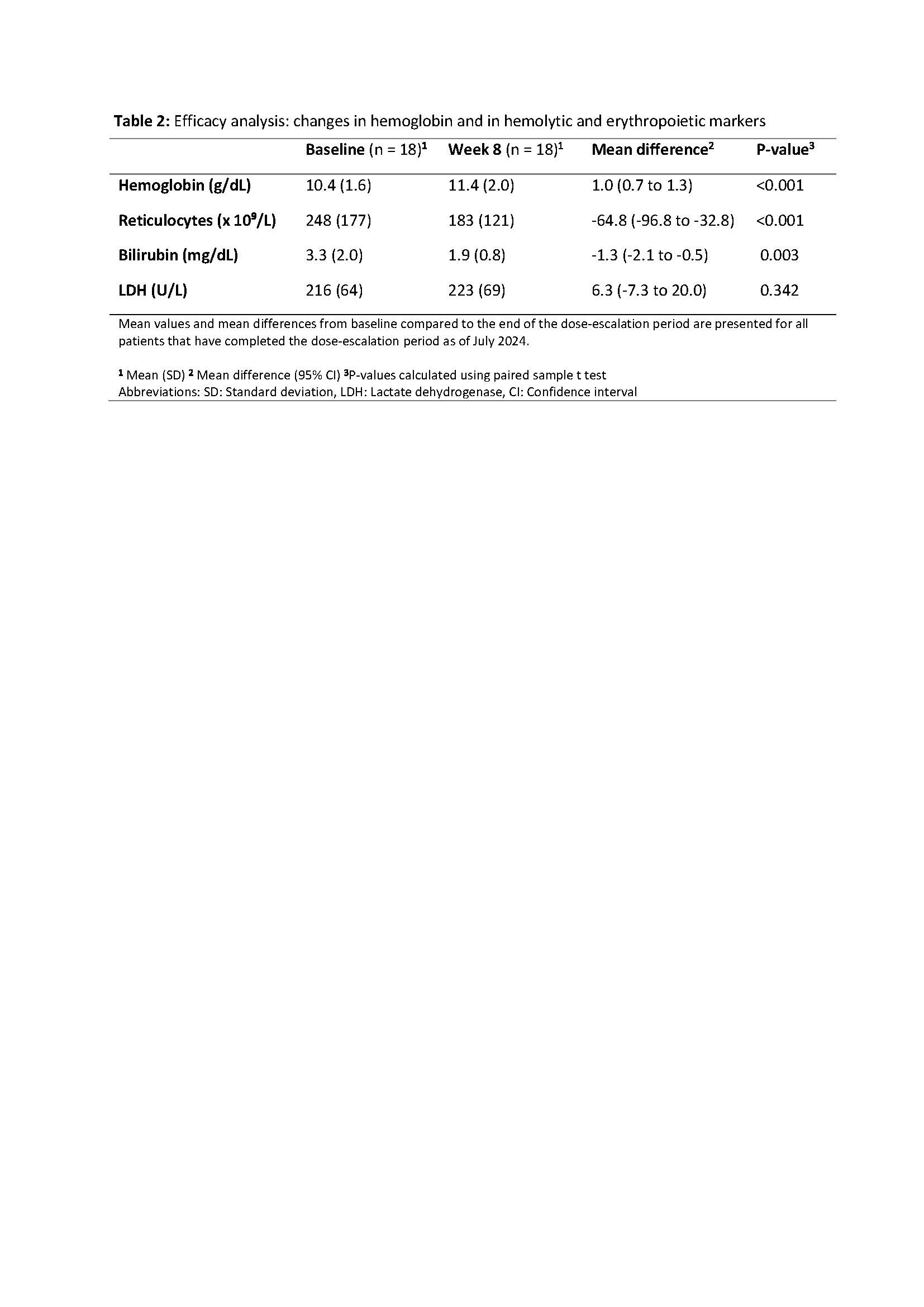
Here, we report data from all patients who completed the 8-week dose-escalation period, during which subjects initially received 50 mg of mitapivat twice daily (BID) for 4 weeks, followed by 100 mg BID. The primary endpoint was safety, as assessed by the occurrence of treatment-emergent adverse events (TEAEs). Secondary endpoints included efficacy, defined as a Hb response of ≥1 g/dL compared to baseline, as well as changes in hemolytic and erythropoietic markers.
As of July 2024, the study has enrolled 24 patients. Of these, 18 patients (12 HS, 4 CDA II and 2 dehydrated hereditary stomatocytosis) completed the dose-escalation period, with a median age of 45 (range 24-79) years and 8/18 (44%) patients being female. Safety analyses revealed mostly (49/50) mild (grade 1-2) TEAEs, with headache and insomnia being the most frequently reported. One serious adverse event (grade 3) occurred, which was unrelated to treatment. Efficacy analyses showed a mean increase in Hb concentration of 1.0 g/dL (standard deviation ± 0.7 g/dL, p < 0.001), with 11/18 (61%) patients reaching the efficacy endpoint of ≥1 g/dL increase (10 HS and 1 CDA II). Analyses of erythropoietic and hemolytic markers showed a significant mean decrease in reticulocytes and total bilirubin, with no significant change in LDH.
In the dose-escalation period of this phase 2 trial, mitapivat has shown initial improvements in hemoglobin and hemolytic markers, with a safety profile consistent with that observed in previous clinical trials.